Bioavailability and Bioequivalence Studies
Introduction
- It is essential to ensure uniformity in standards of quality, efficacy & safety of Pharmaceutical products
- Reasonable assurance is to be provided that various products containing the same active ingredient, marketed by different licensees are clinically equivalent & interchangeable
- Release of an active substance should be known & reproducible
- Both Bioavailability & Bioequivalence focus on the release of the drug substance from its dosage form & subsequent absorption in circulation
- Similar approaches to measure Bioavailability should be followed in demonstrating Bioequivalence
Bioavailability
- Measurement of the relative amount & rate at which, the drug from the administered dosage form, reaches the systemic circulation & becomes available at the site of action
- Bioavailable fraction (F), refers to the fraction of administered dose that enters the systemic circulation
F = Bioavailable dose/Administered dose
OBJECTIVES OF BIOAVAILABILITY STUDIES
- Primary stages of development of a suitable dosage for a new drug entity.
- Development of a new formulation of the existing drugs.
- Control of quality of a drug product during the early stages of marketing in order to determine the influence of processing factors, storage and stability on drug absorption.
- Useful in determining the safety and efficacy of the drug product.
Concept of Equivalents
- Bioequivalence
- Pharmaceutical equivalent/alternative of the test product,
- when administered at the same molar dose,
- has the rate and extent of absorption
- not statistically significantly different from that of the reference product
- Pharmaceutical equivalents
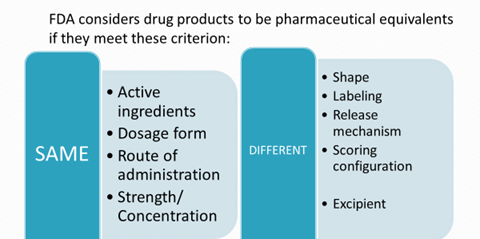
- Pharmaceutical alternatives
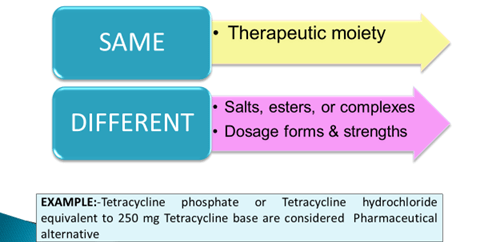
- Therapeutic equivalence – FDA classifies those products as therapeutically equivalent which
- Same active substance or therapeutic moiety
- Clinically show the same efficacy & safety profile
Reference Product
- Identified by the Regulatory Authorities as “Designated Reference Product”
- Usually the Global Innovator’s Product
- Protected by a patent
- Marketed under manufacturers brand name
- Clinical efficacy & safety profile is well documented in extensive trials
- All generics must be Bioequivalent to it
- In India, CDSCO may approve another product as a Reference product
Generic Drug
- Drug product which is identical or bioequivalent to Brand/ Reference drug in: Active ingredient (s), Route of administration, Dosage form, Strength, Indications, Safety
- May have different:
- Inactive ingredients, Colour, Shape
- Almost half of the drugs in the market have Generics
- An Abbreviated New Drug Application (ANDA) is an application for a U.S. generic drug approval for an existing licensed medication or approved drug

Fundamental Bioequivalence Assumption
When a generic drug is claimed bioequivalent to a Reference drug, it is assumed that they are therapeutically equivalent
Bioequivalence Background
- Using bioequivalence as the basis for approving generic copies in the US
“Drug Price Competition and Patent Term Restoration Act of 1984,” also known as the Waxman-Hatch Act
- Created Generic Industry & ↑ their availability
- Most successful legislation
- Benefited Brand & Generic firms
- Generic firms→ Rely on findings of safety & efficacy of Innovator drug after Patent expiration
- Innovator firms→ Patent extensions of 5yrs to make up for time lost while their products were going through FDA’s approval process
Indian Legislation
- In India, CDSCO provides “Guidelines for Bioavailability &
Bioequivalence Studies” mentioned in Schedule Y
- As per the Drugs & Cosmetics Rules (IInd Amendment) 2005, all bioavailability and bioequivalence studies should be conducted in accordance with these Guidelines
The requirement of BA & BE Studies
- For IND/NDAs:
To establish equivalence between:
- Early & late clinical trial formulations
- Formulations used in clinical trial & stability studies
- Clinical trial formulations & to-be-marketed drug product
- Any other comparisons, if appropriate
- ANDA for a generic drug product
- Change in components, composition, &/or manufacturing process
- Change in dosage form (capsules to tablet)
Objectives of BA & BE Studies
- Development of suitable dosage form for a New Drug Entity
- Determination of the influence of excipients, patient-related factors & possible interactions with other drugs
- Development of new drug formulations of existing drugs
- Control of quality of drug products, the influence of → processing factors, storage & stability
- Comparison of availability of a drug substance from a different form or same dosage form produced by different manufacturers
When is Bioequivalence not necessary (Biowaivers)?
- Aqueous parental Solution; a same active substance with the same concentration, same excipient
- Oral Solution; a same active substance with the same concentration, excipient not affecting GI transit or absorption
- Gas
- Powder for reconstitution as a solution; meets criterion (a) or (b)
- Otic/Ophthalmic/Topical Solution; a same active substance with the same concentration, same excipient
- Inhalational Product/ Nasal Spray; administered with or w/o same device as reference product; prepared as an aqueous solution; a same active substance with the same concentration, same excipient

Orange Book
- All FDA approved drugs listed (NDA’s, ANDA’s & OTC’s)
- Expiration of patent dates
- Drug, Price and Competition Act (1984) – FDA required to publish Approved Drug Products with Therapeutic Equivalence & Evaluations
Methods used to assess Equivalence
- Pharmacokinetic Studies
- Pharmacodynamic Studies
- Comparative Clinical Studies
- Dissolution Studies
Pharmacokinetic Studies
Subject selection
- Healthy adult volunteers
- Age: 18-45 yrs
- Age/Sex representation corresponding to a therapeutic & safety profile
- Weight within normal limits→ BMI
- Women: Pregnancy test prior to 1st & last dose of study; OC pills C/I
- Drug use intended in Elders (Age >60yrs)
- Teratogenic Drugs→ Male volunteers
- Highly toxic drugs: Patients with the concerned disease (stable) eg. Cancer
Exclusion Criteria
- H/o allergy to test drug
- H/o liver or kidney dysfunction
- H/o jaundice in the past 6 months
- Chronic diseases eg. Asthma, arthritis
- Psychiatric illness
- Chronic smoker, alcohol addiction, drug abuse
- Intake of enzyme modifying drug in the past 3 months
- Intake of OTC/Prescription drugs past 2 weeks
- HIV positive
- BA & BE studies in the past 3 months
- H/o bleeding disorder
Selection of Number of Subjects
- The sample size is estimated by:
- Pilot experiment
- Previous studies
- Published data
- Significance level desired, usually 0.05
- Power of the study, normally 80% or more
- Expected deviation (Δ) from the reference product, as compatible with BE
- If no data available, reference ratio of 0.95 (Δ = 5%) used

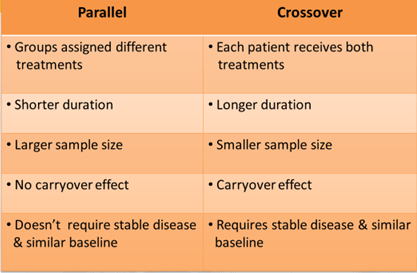
Study Design
- Good experimental design enhances the power of the study
- Depends on: question to be answered, nature of reference drug/ dosage form, the benefit-risk ratio
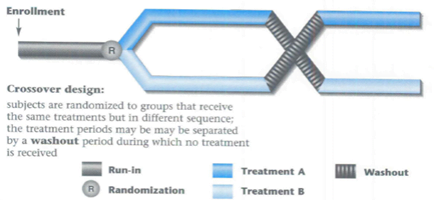
- As far as possible, the study should be of crossover design & suitably randomized
- Ideal design: Randomized two-period, two-sequence, Crossover design with an adequate washout period
- If the half-life is long: Parallel design
- For highly variable drugs: Replicate design
- Two-Period Crossover Design
- 2 formulations, even number of subjects, randomly divided into 2 equal groups
- First period, each member of one group receive a single dose of the test formulation; each member of the other group receive the standard formulation
- After a wash period (5 half-lives), in the second period, each member of the respective groups will receive an alternative formulation & the experiment will be repeated.
Parallel-Group Design
- Even the number of subjects in two groups
- Each receives a different formulation
- No washout necessary
- For drugs with a long half-life
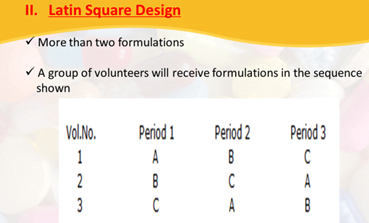
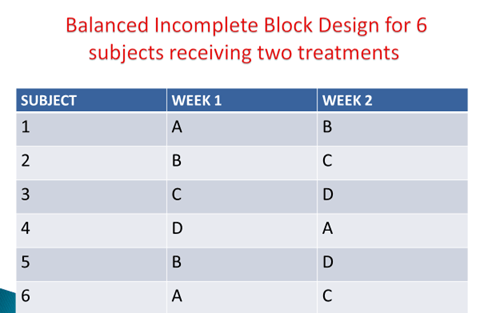
Genetic Phenotyping
- The drug is known to be subject to genetic polymorphism
- Cross-over design→ Safety & Pharmacokinetic reasons
- All Parallel group design
- Indian population:
- Captures genetic diversity of the world
- Forms continuum of the genetic spectrum
- >1000 medically relevant genes
- Diverse patient/ volunteer pool for conducting BA & BE studies
Characteristics to be measured
- Accessible biological fluids like blood, plasma &/or serum to indicate the release of the drug substance from the drug product into the systemic circulation
- Mostly: Active drug substance
- Active / Inactive metabolite may be measured in cases of:
- The concentration of drug too low, Limitation of the analytical method, Unstable drug, Drug with a very short half-life, Pro-drugs
- Excretion of drug & its metabolites in urine→ Non-linear kinetics
- Drugs that are not absorbed systemically from the site of application surrogate marker needed for BA & BE determination
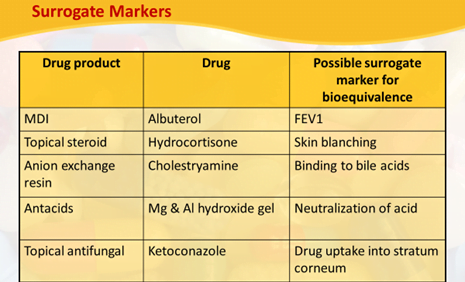
Blood Sampling points/ Schedule
A single-dose study of an immediate release product:
- For at least three
elimination half-lives (cover >80% of AUC)
- Absorption phase: 3-4 points
- Around Tmax: 3-4points
- During elimination: 4 points
- Intervals not longer than the half-life of the drug
- If urine tested, collect it for at least 7 half-lives
Parameters to be measured
- Pharmacokinetic Parameters measured are:
- Cmax
- Tmax
- AUC0-t
- AUC0-∞
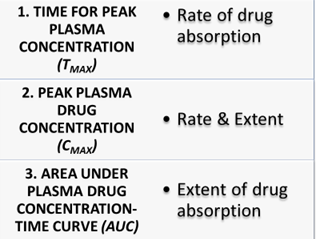
Fasting & Fed State Conditions
Fasting Conditions:
- Single-dose study:
- Overnight fast (10 hrs) and subsequent fast of 4 hrs
- Multiple-dose study:
- Two hours fasting before and after the dose
Fed State Studies
- Required when:
- The drug recommended with food
- Modified release product
- Assessment of Cmax and Tmax difficult with fasting state study
- Requires consumption of high-fat food, 15 minutes before dosing
- Provide 950-1000 kcals
- Fat- 50%, Proteins 15-20%, Carbohydrate- 30-35%
- Ethnic & cultural variation considered
- Specified in protocol
Statistical Evaluation
- Cmax & AUC analyzed using ANOVA
- Tmax analyzed by non-parametric methods
To establish BE:
- The calculated 90% CI for Cmax & AUC, should fall within range: 80-125% (Range of Bioequivalence)
- Non-parametric data 90% CI for Tmax should lie within a clinically acceptable range
Documentation
- Signed detailed protocol
- Approval by Ethics Committee
- Participant Information sheet
- Informed Consent Form (ICF)
- Case Record Form (CRF)
- Undertaking by investigator
- CV of investigator
- Randomization chart
- Laboratory certification
- Analytical method validation details
- Chromatograms of all volunteers including any aberrant ones
- Tabulated Raw Data of volunteers
Maintenance of Records & Retention of Study Samples
- All Records of in vivo tests on any marketed batch of a drug product should be maintained by the Sponsor
for at least 2 years after the expiry date of the batch
- All Drug samples to be retained for a period of at least 3 years after the conduct of the study
OR
1year after the expiry of the batch [Stored in conditions consistent with the product labeling]