Pharmacotherapy of Pulmonary Hypertension
- Definition- Pulmonary Hypertension (PH) is defined as an increase in mean pulmonary arterial pressure (PAPm) ≥25 mmHg at rest as assessed by right heart catheterization (RHC).
- Normal (PAPm) – 14 ± 3 mmHg
- PAPm of 21 – 24 mmHg – Carefully followed
Classification – Based on Etiology
- Group 1 – Pulmonary Arterial Hypertension Idiopathic Heritable à BMPR2 mutationDrugs and toxins induced Associated with: Connective tissue diseaseHIV Infection Portal hypertension Congenital heart disease Schistosomiasis
- Group 2 – PH Due to Left Heart Disease
- Group 3 – Pulmonary Hypertension Due to Lung Disease and/or Hypoxia
- Group 4 – Chronic Thromboembolic Pulmonary Hypertension (CTEPH)
- Group 5 – Pulmonary Hypertension with Unclear Multifactorial Mechanisms
- PAH
- Affects 15-50 people/million – Globally
- Affects all ages; most prevalent in 4th & 5th decades of life
- Higher prevalence in females than males (2-3:1 ratio)
- PH due to Left Heart Disease – most common cause of PH
Pathophysiology
- Pulmonary vascular remodelling
- Sustained pulmonary vasoconstriction
- In- situ thrombosis
- Pulmonary vascular wall stiffening
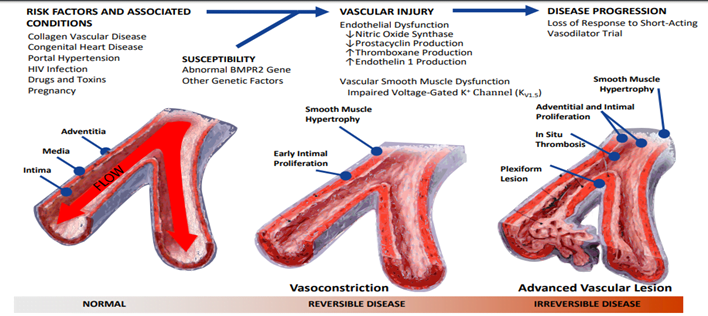
Pathophysiology & Targets

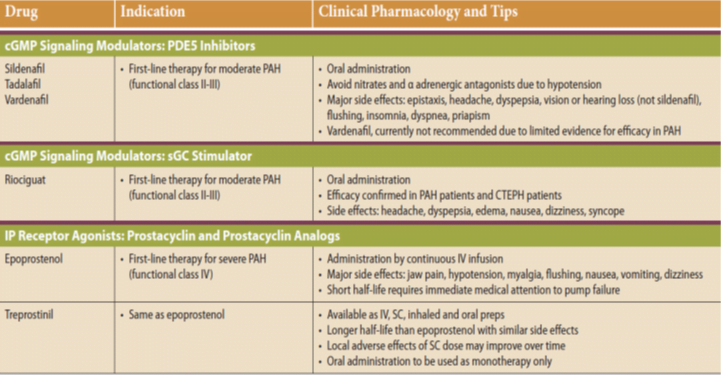
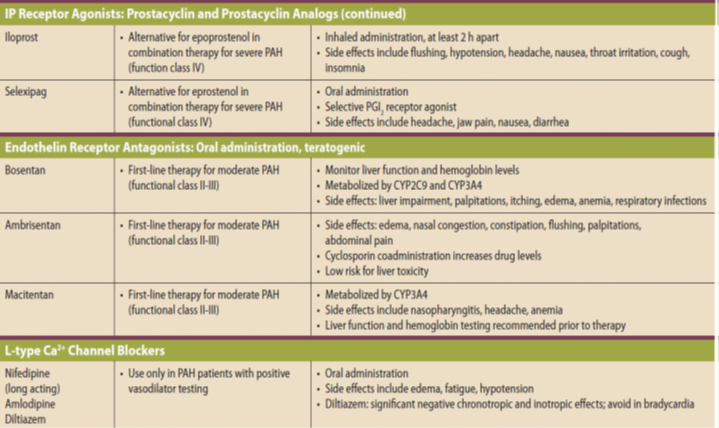
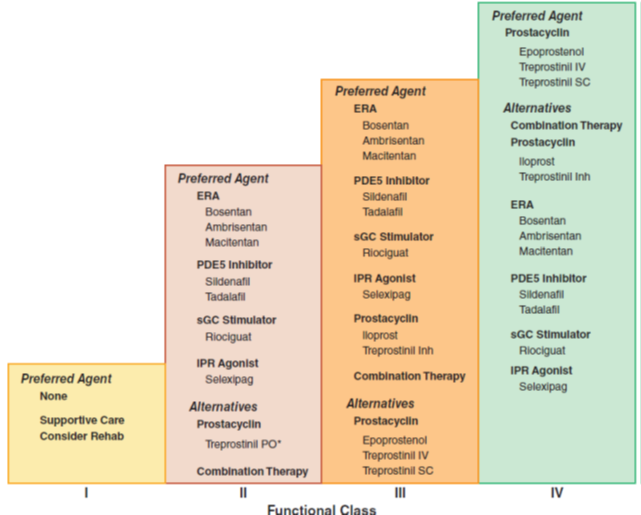
WHO Functional Classification of Pulmonary Hypertension
Four functional classes have been defined for PAH:
(I) no symptoms or functional limitation;
(II) slight limitation of physical activity;
(III) marked limitation of physical activity; and
(IV) symptoms with any activity or at rest.